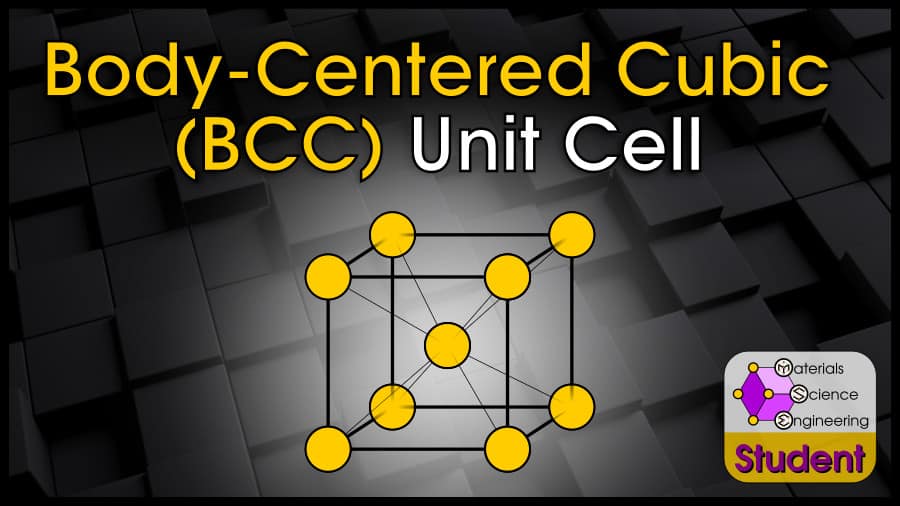
The Body-Centered Cubic (BCC) crystal structure is one of the most common ways for atoms to arrange themselves in metals. The BCC crystal structure is based on the Bravais lattice of the same name, with 1 atom per lattice point at each corner of the cube and the center of the cube. BCC is nearly close-packed and has great stability because of its high number of nearest neighbors and next-nearest neighbors. The body-centered cubic cell belongs to space group #229 or  , Strukturbericht A2, and Pearson symbol cI2. W is the prototype for BCC.
, Strukturbericht A2, and Pearson symbol cI2. W is the prototype for BCC.

The Body-Centered Cubic (BCC) unit cell can be imagined as a cube with an atom on each corner, and an atom in the cube’s center. It is one of the most common structures for metals. BCC has 2 atoms per unit cell, lattice constant a = 4R/√3, Coordination number CN = 8, and Atomic Packing Factor APF = 68%.
Don’t worry, I’ll explain what those numbers mean and why they’re important later in the article. For now, let’s talk about which materials actually exist as body-centered cubic.
Outline
- Common Examples of Body-Centered Cubic Materials
- Body-Centered Cubic Coordination Number
- Body-Centered Cubic Lattice Constants
- Body-Centered Cubic Atomic Packing Factor
- Primitive Body-Centered Cubic Unit Cell
- Interstitial Sites in Body-Centered Cubic
- Slip Systems in Body-Centered Cubic
- Final Thoughts
- References and Further Reading
Common Examples of Body-Centered Cubic Materials
Since BCC is one of the most common crystal structures, there are many examples to choose from!
Lithium, sodium, potassium, vanadium, chromium, iron, rubidium, niobium, molybdenum, cesium, barium, europium, tantalum and tungsten all have the BCC crystal structure. This list is not comprehensive, and BCC can also appear as high temperature/pressure phases, in solidified nonmetals, or in alloys.

BCC metals have a ductile-to-brittle transformation temperature (DBTT), which means that they are brittle at lower temperatures and ductile at higher temperatures. So, some BCC metals are ductile at room temperature, and others are brittle.
If you are interested in why BCC metals have different properties than FCC metals (another common structure), you should check out this article.
BCC metals also tend to have a high melting point, because they have a large number of nearest neighbors and next-nearest neighbors.
Body-Centered Cubic Coordination Number
Coordination Number (CN) is the number of nearest neighbors that each atom has.
In a body-centered cubic crystal, each atom has 8 nearest neighbors (NN). That is not the maximum (which is 12, found in close-packed structures), but BCC has such high stability because of its next-nearest neighbors.
There are 6 next-nearest neighbors, but these are only 15% farther away than the nearest-neighbors. In some sense, this allows BCC crystals to act as though they have 14 bonds, which is higher than the maximum of 12. This may be one reason that most high melting point metals have a BCC structure.

The nearest neighbor distance is  , and the next-nearest neighbor distance is
, and the next-nearest neighbor distance is  (or about
(or about  ).
).
Body-Centered Cubic Lattice Constants
The body-centered cubic lattice is a cube with an atom on each corner and another in the volumetric center of the cube. Using the hard sphere model, which imagines each atom as a discrete sphere, the BCC crystal has each atom touch along the body diagonal of the cube.
That means that the body diagonal has a length of  , which works out to that the lattice parameter
, which works out to that the lattice parameter  , or side length of the cube, has a length of .
, or side length of the cube, has a length of .

If you wanted to describe the face-centered cubic crystal with math, you would describe the cell with the vectors



These are actually primitive vectors, which you can read about in the section below.
Body-Centered Cubic Atomic Packing Factor
The Atomic Packing Factor (APF) is essentially the density of the unit cell. Since we use the hard sphere model, each point inside the cell is either part of an atom, or part of the void.
APF is the

APF is basically the fraction of atoms to void. For a full article explaining APF, check out this link.
The total volume of the unit cell is just the volume of a cube. The cube side length is a, the volume is  .
.
Now we need to count how many atoms are in each unit cell. It may look like there are 9 atoms because there are 8 corners and 1 in the center, but actually the cell only intersects portions of those corner atoms.
If you count the portion of the atoms in the cell, 1/8th of each atom would count, and there are 8 corner atoms.  , so there is one full corner atom. There is also a full atom in the center of the cube.
, so there is one full corner atom. There is also a full atom in the center of the cube.
Thus, there are 2 atoms per unit cell.
The volume of a sphere is  . We previously established that the volume of the whole cube is , and since
. We previously established that the volume of the whole cube is , and since  , the volume of the cube is
, the volume of the cube is  .
.
Now that we’ve written everything in terms of the radius of an atom, you can see that every face-centered cubic crystal will have the same packing factor regardless of the actual element.


As you can see, body-centered cubic crystals have 68% packing. That’s almost as much as a close-packed crystal (which has APF = 74%).
Primitive Body-Centered Cubic Unit Cell
Advanced topic, click to expand!
The conventional BCC cell that I have shown you is a conventional unit cell, not a primitive unit cell. This conventional cell has advantages because it is highly symmetric and easy for humans to understand.
However, when dealing with mathematical descriptions of crystals, it may be easier to describe the unit cell in the smallest form possible. The smallest possible unit cell is called the primitive cell. If you are interested in primitive cells, you can read all about them in this article.
The BCC primitive cell looks like this:

Here are the primitive vectors for the BCC unit cell.
Interstitial Sites in Body-Centered Cubic
Interstitial sites are the spaces inside a crystal where another kind of atom could fit. You can read all about interstitial sites in this article, but BCC has two types of interstitial sites: octahedral and tetrahedral.
BCC has 6 octahedral sites, which means that a small interstitial atom could fit in 6 positions such that it is equally surrounded by 6 FCC lattice atoms.

These octahedral interstitial atoms can be  size.
size.
BCC also has 12 tetrahedral sites, which means that a small interstitial atom could fit in 12 positions such that it is equally surrounded by 4 FCC lattice atoms.
These tetrahedral atoms can be  size.
size.
Slip Systems in Body-Centered Cubic
Advanced topic, click to expand!
Slip systems are the way that atoms slide past each other when deforming. Slip systems determine many mechanical properties of materials, and is the main reason why a material will be ductile or brittle.
To understand slip system directions, you will need to be familiar with Miller Indices notation. Look for this article in the future on our website!
BCC crystals have 48 slip systems but only 5 are independent (5 is the minimum requirement for ductility). However, the 5 independent slip systems may not be activated at all temperatures, so some BCC metals are ductile while others are brittle.
The 48 BCC slip systems are {110}<111>, {112}<111>, and {123}<111> in order of ease of activation. I’d recommend reading “Kelly & Knowles, Crystallography and Crystal Defects” if you want to prove this, and if you want to prove that only 5 of these are independent.
Further complicating BCC ductility: these slip systems may not always be active. Unlike FCC slip systems, where planes are truly close-packed, BCC slip planes don’t have atoms touching. They need thermal energy for atoms to overcome this extra distance and slip.

Depending on the element, the thermal energy to activate the slip systems will be different. At room temperature, BCC iron has all 5 slip systems activated, which is why it is ductile. If you brought iron below room temperature, however, there would not be enough thermal energy to activate all the slip systems.
This phenomenon is called the ductile-to-brittle transformation temperature (DBTT). All BCC metals have a DBTT.

Final Thoughts
The Body-Centered Cubic (BCC) crystal structure is important because it is extremely common in metals, and results in interesting properties like the ductile-to-brittle transformation temperature (DBTT) and high melting points.
BCC is nearly close-packed and has an APF of 0.68. Many of its properties arise because the next-nearest neighbors bond nearly as powerfully as the nearest neighbors.
Here is a summary chart of all BCC crystal properties:
| Crystal Structure | Body-Centered Cubic (BCC) |
| Unit Cell Type | Cubic |
| Relationship Between Cube Edge Length a and the Atomic Radius R | a = 4R/√3 |
| Close-Packed Structure | No |
| Atomic Packing Factor (APF) | 68% |
| Coordination Number | 8 |
| Number of Atoms per Unit Cell | 2 |
| Number of Octahedral Interstitial Sites | 6 |
| Number of Tetrahedral Interstitial Sites | 12 |
| Size of Octahedral Voids | r = 0.155 R |
| Size of Tetrahedral Voids | r = 0.291 R |
References and Further Reading
If you want to know more about the basics of crystallography, check out this article about crystals and grains.
If you weren’t sure about the difference between crystal structure and Bravais lattice, check out this article.
I also mentioned atomic packing factor (APF) earlier in this article. This is an important concept in your introductory materials science class, so if you want a full explanation of APF, check out this page.
If you’re interested in advanced crystallography or crystallography databases, you may want to check out the AFLOW crystallographic library.
For a great reference for all crystal structures, check out “The AFLOW Library of Crystallographic Prototypes.”
Single-Element Crystal Structures and the 14 Bravais Lattices
If you want to learn about specific crystal structures, here is a list of my articles about Bravais lattices and some related crystal structures for pure elements. Body-Centered Cubic is one of these 14 Bravais lattices and also occurs as a crystal structure.
1. Simple Cubic
2. Face-Centered Cubic
2a. Diamond Cubic
3. Body-Centered Cubic
4. Simple Hexagonal
4a. Hexagonal Close-Packed
4b. Double Hexagonal Close-Packed (La-type)
5. Rhombohedral
5a. Rhombohedral Close-Packed (Sm-type)
6. Simple Tetragonal
7. Body-Centered Tetragonal
7a. Diamond Tetragonal (White Tin)
8. Simple Orthorhombic
9. Base-Centered Orthorhombic
10. Face-Centered Orthorhombic
11. Body-Centered Orthorhombic
12. Simple Monoclinic
13. Base-Centered Monoclinic
14. Triclinic
Other articles in my crystallography series include:
Introduction to Bravais Lattices
What is the Difference Between “Crystal Structure” and “Bravais Lattice”
Atomic Packing Factor
How to Read Miller Indices
How to Read Hexagonal Miller-Bravais Indices
Close-Packed Crystals and Stacking Order
Interstitial Sites
Primitive Cells
How to Read Crystallography Notation
What are Point Groups
List of Point Groups
What are Space Groups
List of Space Groups
The 7 Crystal Systems